A regulatory submission is not a filing—it is an argument. At Boulder Solution, we don’t just compile data; we architect defensible evidence. Most delays in the 510(k) or CE Mark process stem from fragmented data or Substantial Equivalence arguments that don’t hold up under FDA or Notified Body scrutiny.
Regulatory Pathways & Submission Types
Our Submission Expertise Covers:

Submission Documentation:
- Substantial Equivalence (SE) Determination
- Predicate Device Comparison
- Indications for Use Statement
- Device Description & Specifications
- Performance Testing & Bench Testing
- Biocompatibility Testing (ISO 10993)
- Sterilization Validation (ISO 11135, ISO 11137)
- Electrical Safety Testing (IEC 60601)
- Software Documentation (IEC 62304)
- Usability Testing (IEC 62366 / Human Factors)
- Clinical Evaluation Reports (CER)
- Risk Management Files (ISO 14971)
- Labeling & Instructions for Use (IFU)
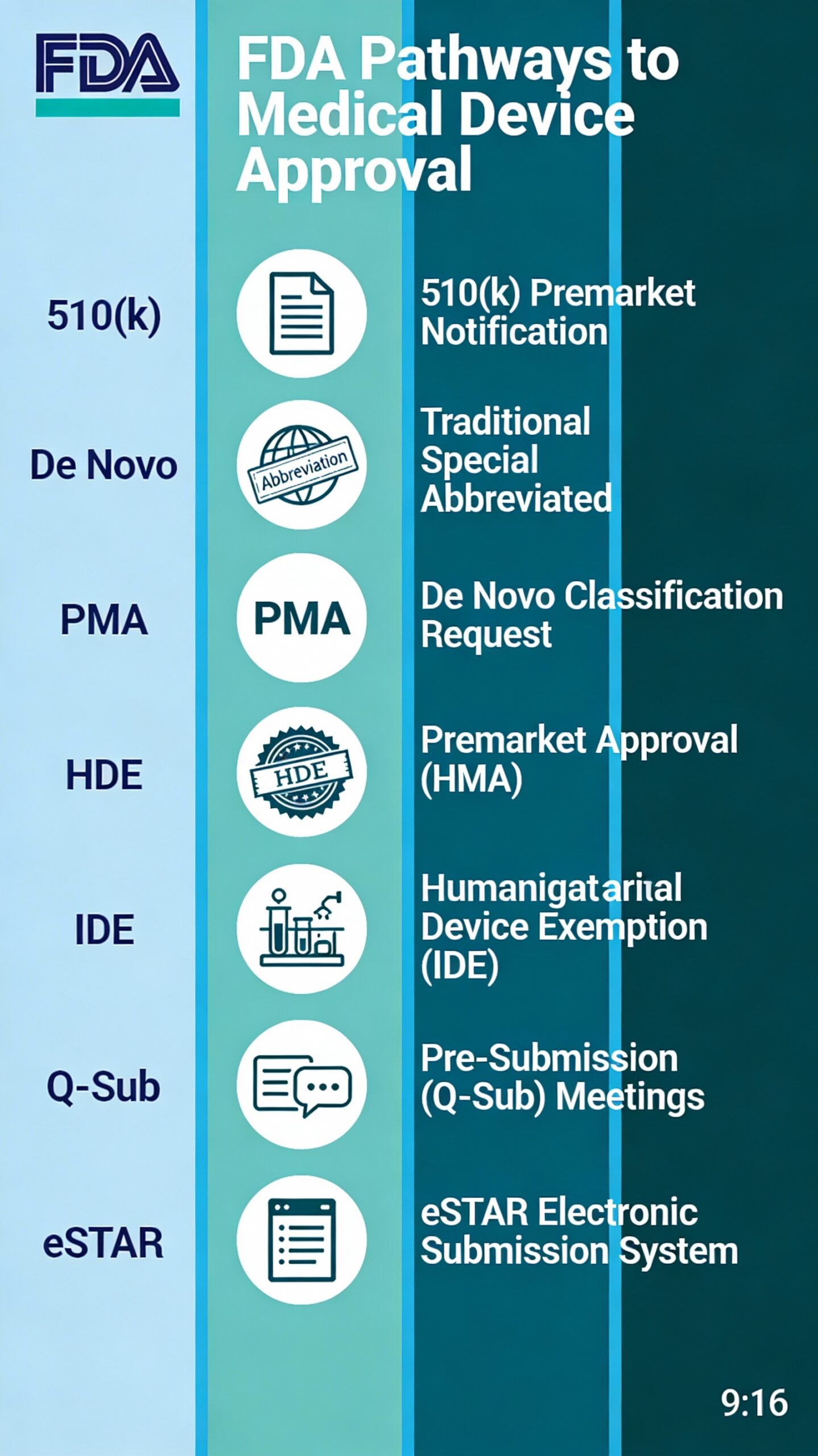
US FDA Pathways:
- 510(k) Premarket Notification (Traditional, Special, Abbreviated)
- De Novo Classification Request
- Premarket Approval (PMA)
- Humanitarian Device Exemption (HDE)
- Investigational Device Exemption (IDE)
- Pre-Submission (Q-Sub) Meetings
- eSTAR Electronic Submission System
EU & International Pathways:
- CE Mark Technical File (EU MDR 2017/745)
- CE Mark Transition from MDD to MDR
- UKCA Mark (UK Conformity Assessed)
- Health Canada Medical Device License (MDL)
- TGA (Australia) Conformity Assessment
- PMDA (Japan) Marketing Approval
- ANVISA (Brazil) Registration
Testing & Validation Support:
- Design Verification & Validation (V&V)
- Performance Testing Protocols
- Animal Testing & Preclinical Studies
- Clinical Trial Design & Monitoring
- Statistical Analysis Plans
- Comparative Effectiveness Studies
Post-Market Requirements:
- Post-Market Surveillance (PMS) Plans
- Periodic Safety Update Reports (PSUR)
- Medical Device Reporting (MDR)
- Summary of Safety & Clinical Performance (SSCP)
- Post-Approval Studies