Medical Device Approval: A Strategic Guide to US & EU Market Access
Founders of medical device startups and biomedical engineers often face a critical, early challenge: navigating the intricate regulatory landscape. Bringing a medical device to market, whether in the United States or the European Union, demands rigorous adherence to regulatory processes and stringent quality controls.
This guide aims to demystify these requirements by detailing:
-
- The foundational importance of device classification.
-
- Distinct FDA approval pathways (510(k) vs. PMA).
-
- The nuances of CE Mark conformity under EU MDR/IVDR.
-
- The indispensable role of ISO 13485 certification.
Medical Device Approval: A Strategic Guide to US & EU Market Access
Founders of medical device startups and biomedical engineers often face a critical, early challenge: navigating the intricate regulatory landscape. Bringing a medical device to market, whether in the United States or the European Union, demands rigorous adherence to regulatory processes and stringent quality controls. This guide aims to demystify these requirements.
It explains the foundational importance of device classification, the distinct FDA approval pathways like 510(k) clearance and Premarket Approval (PMA), the nuances of CE Mark conformity under EU MDR/IVDR, and the indispensable role of ISO 13485 certification.
Our objective is to provide a comprehensive, go-to resource. We detail how classification impacts regulatory burden, outline FDA documentation and testing expectations for different submissions, explain how notified bodies assess conformity for CE marking, and demonstrate how a certified quality management system (QMS) facilitates both U.S. and EU market access.
The discussion focuses on practical steps, potential timelines, common pitfalls, and actionable strategies, empowering teams to plan studies, meticulously prepare technical documentation, manage post-market obligations, and effectively engage with regulatory authorities. This isn’t just theory; it’s direct advice from a consultant who has spent over 16 years in regulatory affairs, witnessing firsthand the critical junctures and potential missteps.
Understanding FDA Medical Device Classifications and Their Regulatory Impact
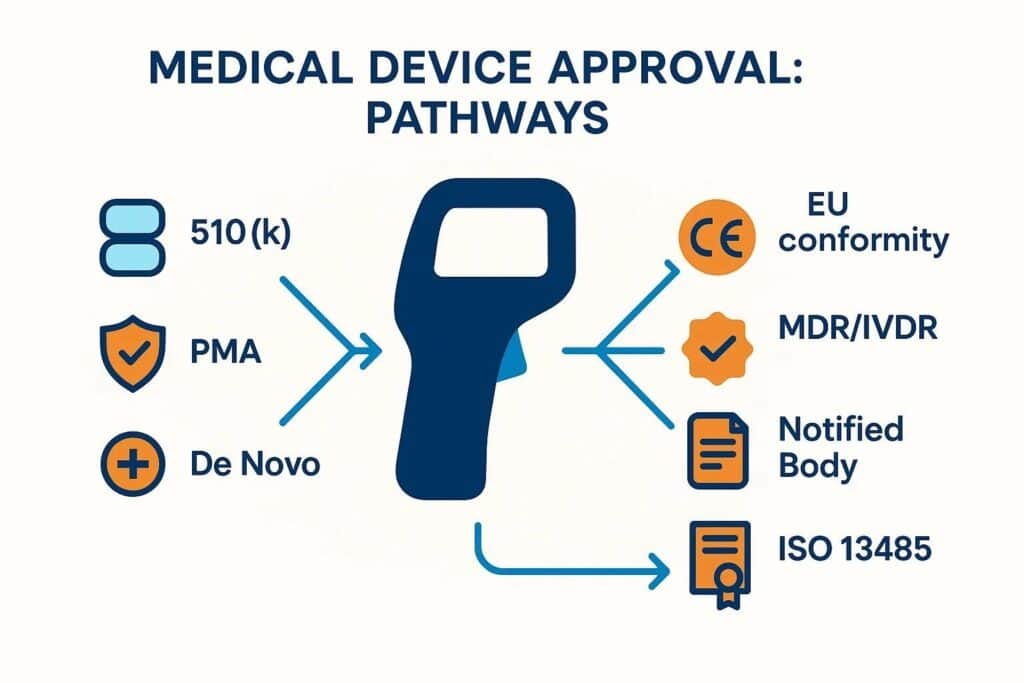
Device classification is the cornerstone of regulatory strategy in the U.S. It assigns a risk tier—Class I, II, or III—directly influencing the scope of applicable regulatory controls and the specific approval process. This early determination matters significantly. It dictates the required evidence burden, the necessity of general or special controls, or ultimately a PMA, and the projected timelines.
Devices deemed lower risk typically rely on general controls and may even be exempt from premarket notification. Conversely, higher-risk devices demand far more rigorous data and comprehensive review. Misclassifying a device can lead to substantial delays and wasted resources. An accurate initial assessment informs everything: biocompatibility testing, performance evaluation, the need for clinical trial data, and even labeling requirements.
Consider this simplified classification matrix:
| Device Class | Risk Level | Typical Examples | Common Regulatory Pathways |
| Class I | Low | Tongue depressor, elastic bandage | General controls; many exempt from premarket notification |
| Class II | Moderate | Infusion pumps, surgical lasers | 510(k) clearance or De Novo if no predicate; special controls |
| Class III | High | Implantable defibrillators, stents | PMA; rigorous clinical and manufacturing evidence |
This matrix underscores why a precise risk assessment at the outset saves both time and significant capital. It aligns your testing, technical documentation, and submission strategy with the FDA’s expectations.
Defining and Regulating Class I, II, and III Devices
Class I devices carry minimal risk. They are primarily subject to general controls, which include requirements for proper labeling, facility registration, and basic quality management system adherence. Many Class I products are exempt from premarket notification, though they must still meet essential manufacturing and labeling obligations.
Class II devices present a moderate risk. These devices require special controls in addition to general controls. Manufacturers typically pursue a 510(k) submission, demonstrating substantial equivalence to an existing legally marketed predicate device. If no suitable predicate exists, a De Novo classification request might be an option.
Class III devices represent the highest risk category. These devices are often life-sustaining, life-supporting, implanted, or pose a potentially unreasonable risk of illness or injury. For these, a PMA is typically mandated. This process demands extensive clinical data to definitively demonstrate the device’s safety and efficacy, and it operates under the most stringent regulation.

Understanding these definitions early enables teams to select appropriate standards, develop robust testing protocols, and plan pre-submission interactions effectively. This mitigates review deficiencies and streamlines the overall regulatory compliance effort.
This precise regulatory framing then directly leads to pathway selection: 510(k), De Novo, IDE-supported clinical studies, or PMA submission. The choice hinges on specific device features, its intended use, and the availability of suitable predicates.
Implications of Device Classification on Regulatory Pathways
Device classification directly dictates the appropriate regulatory pathway and the intensity of required clinical evidence. It profoundly shapes timelines, ranging from months for many 510(k)s to several years for PMA reviews and pivotal trials.
For instance, an electrical monitor, if based on a clear predicate, could achieve 510(k) clearance with bench testing and limited clinical performance evaluation. However, a novel implantable device lacking a predicate would almost certainly require a De Novo or PMA pathway, necessitating substantive clinical investigation under an Investigational Device Exemption (IDE).
Risk controls further influence labeling, post-market surveillance, and post-approval study obligations. This, in turn, impacts manufacturing controls and corrective and preventive action (CAPA) planning. Consequently, early classification decisions should drive a comprehensive regulatory plan.
This plan must accurately sequence verification and validation testing, define clinical study design, and outline quality management system activities. Such an integrated approach helps avoid costly rework and aligns product development with FDA expectations.
Explicit pathway planning reduces uncertainty when developmental changes arise. It also provides clear decision points—such as whether to pursue a De Novo request or design an IDE-led trial—that align with business timelines and risk management strategies.
The 510(k) Submission Process: Navigating Market Entry for Moderate-Risk Devices
A 510(k) submission aims to demonstrate that a new medical device is substantially equivalent to a legally marketed predicate device. This pathway allows moderate-risk devices (Class II medical devices) to enter the U.S. market without the full burden of a PMA.
The process operates by aligning the new device’s indications for use, technological characteristics, and performance data with a predicate device. This allows the FDA to reasonably conclude equivalent safety and effectiveness. Fundamentally, this mechanism helps shorten time-to-market for many Class II products, a crucial factor for medical device manufacturers.
Successful 510(k) preparation requires a comprehensive technical file, supported by appropriate bench and, where necessary, clinical evidence, along with clear labeling and a thorough risk analysis. Engaging in pre-submission (Q-Sub) interactions with the FDA can prove invaluable. These early discussions often clarify testing expectations, statistical endpoints, and whether additional clinical data will be necessary, significantly reducing the likelihood of a substantial equivalence denial.
The following element breakdown can assist teams in assigning ownership and planning early document production. This ensures efficient reviewer responses and minimizes review cycles.
| 510(k) Element | Requirement | Typical Owner / Purpose |
| Predicate Device | Demonstrate substantial equivalence | Regulatory lead compares indications and tech characteristics |
| Device Description | Design, components, software, accessories | Engineering provides detailed schematics and specs |
| Performance Testing | Bench, biocompatibility, electrical safety | Test lab delivers validated reports and protocols |
| Sterilization & Shelf-life | Validation and packaging | QA/CMC documents processes and stability data |
| Labeling & Instructions | Intended use and warnings | Regulatory drafts, legal reviews labeling content |
| Summary & Truthful Statements | Executive summary and certifications | Regulatory compiles submission narrative |
Steps and Requirements for a Successful 510(k) Submission
A successful 510(k) submission follows a defined sequence. First, clearly articulate the device’s intended use and identify the most relevant predicate. Then, conduct a thorough risk analysis (e.g., per ISO 14971) and execute all necessary verification and validation (V&V) testing.
This includes bench, software, and biocompatibility tests. Compile a detailed device description, complete labeling, and robust performance data. Manufacturers may consider a pre-submission meeting with the FDA to clarify testing expectations and statistical endpoints. Finally, prepare the submission in FDA-accepted formats, ensuring comprehensive test evidence supports every claim.
Key 510(k) workflow checkpoints include:
1. Define Device & Predicate: Establish clear indications for use; identify the most relevant predicate medical device.
2. Complete Risk Management: Perform risk analysis per ISO 14971, mapping mitigations to specific tests.
3. Execute V&V Testing: Complete bench, software, and biocompatibility testing with traceable protocols.
4. Prepare Labeling & Instructions: Draft clear, compliant labeling and warnings.
5. Submit & Respond: File the 510(k); be ready to address deficiency letters promptly.
These steps directly affect acceptance rates and timelines. Organized evidence and early engagement typically reduce deficiency letters and shorten overall review durations. Maintaining an organized, indexed submission package, anticipating reviewer questions, is crucial.
Common Reasons for 510(k) Submission Failures and Mitigation Strategies
Failures in 510(k) submissions commonly stem from insufficient predicate justification, incomplete or poorly formatted test reports, or a lack of traceability between requirements and test results. Inadequate software documentation for Software as a Medical Device (SaMD), along with labeling that conflicts with intended use statements, also frequently cause issues.
To avoid these pitfalls, ensure each test report contains a clear protocol, defined pass/fail criteria, raw data appendices, and a summary directly linking results to device requirements. A trace matrix, connecting design inputs to verification and validation activities, is not merely good practice—it’s essential. For connected devices or those incorporating AI/ML, document software risk controls and cybersecurity measures diligently, following all relevant FDA guidance.
Statistical underpowering in studies or the absence of clinically relevant performance evaluation metrics can also prompt reviewers to request additional data. Planning sample sizes and endpoints meticulously and in advance can mitigate this risk. Internal technical reviews and, when appropriate, pre-submission meetings, serve as critical checkpoints.
They confirm expectations and reduce the likelihood of a “refusal to accept” or significant deficiency letters. Implementing robust document control and mandatory cross-functional sign-offs before submission establishes a defensible narrative, addressing reviewer expectations and ultimately shortening overall review timelines.
Premarket Approval (PMA): The Rigorous Path for High-Risk Devices
The Premarket Approval (PMA) pathway represents the FDA’s most stringent approval process for medical devices. It is typically reserved for Class III devices, those that are life-sustaining, life-supporting, or pose a significant risk of illness or injury. PMA requires substantial scientific evidence demonstrating a reasonable assurance of safety and effectiveness.
This often includes pivotal clinical trials, extensive CMC (chemistry, manufacturing, and controls) documentation, and a thorough risk-benefit analysis. The PMA process is characterized by an exceptionally rigorous review, often involving an advisory committee evaluation, and formal post-approval commitments. It ensures that high-risk devices meet the highest evidentiary standards before they reach the market.
Medical device manufacturers must carefully plan IDE studies to generate the necessary clinical data. They must also develop robust manufacturing controls in full adherence to 21 CFR Part 820 quality system requirements. This requires preparing extensive modules that cover clinical, nonclinical, and manufacturing data.
Because PMA reviews are considerably more resource-intensive and time-consuming than 510(k) submissions, manufacturers must align clinical strategy, manufacturing scale-up, and supply-chain controls early in development. This proactive approach helps avoid costly delays during regulatory review.
How PMA Differs from 510(k) Clearance
PMA fundamentally differs from 510(k) clearance in several critical aspects: evidence intensity, review depth, and post-approval obligations. PMA demands “valid scientific evidence”—typically from well-designed pivotal studies, often randomized and controlled. It requires highly detailed CMC data and comprehensive risk analyses. In contrast, a 510(k) relies on demonstrating substantial equivalence to a predicate, generally with less rigorous clinical data requirements.
Timelines for PMA submissions are substantially longer. Premarket data collection, multiple FDA inspections (including manufacturing facilities), potential panel reviews, and mandated post-approval studies can extend the process over multiple years. PMA applicants should anticipate deeper scrutiny of manufacturing processes and facilities, stricter device controls, and ongoing market surveillance. This includes explicit post-approval study commitments.
Practically, pursuing a PMA significantly impacts budget, clinical development timelines, and commercialization planning. It often necessitates sustained interaction with the FDA across several milestones: IDE submissions, pivotal trial monitoring, and pre-PMA meetings. This comparison underscores why an early decision regarding the appropriate pathway—510(k), De Novo, or PMA—is critical for clinical planning, managing investor expectations, and ensuring optimal product launch timing.
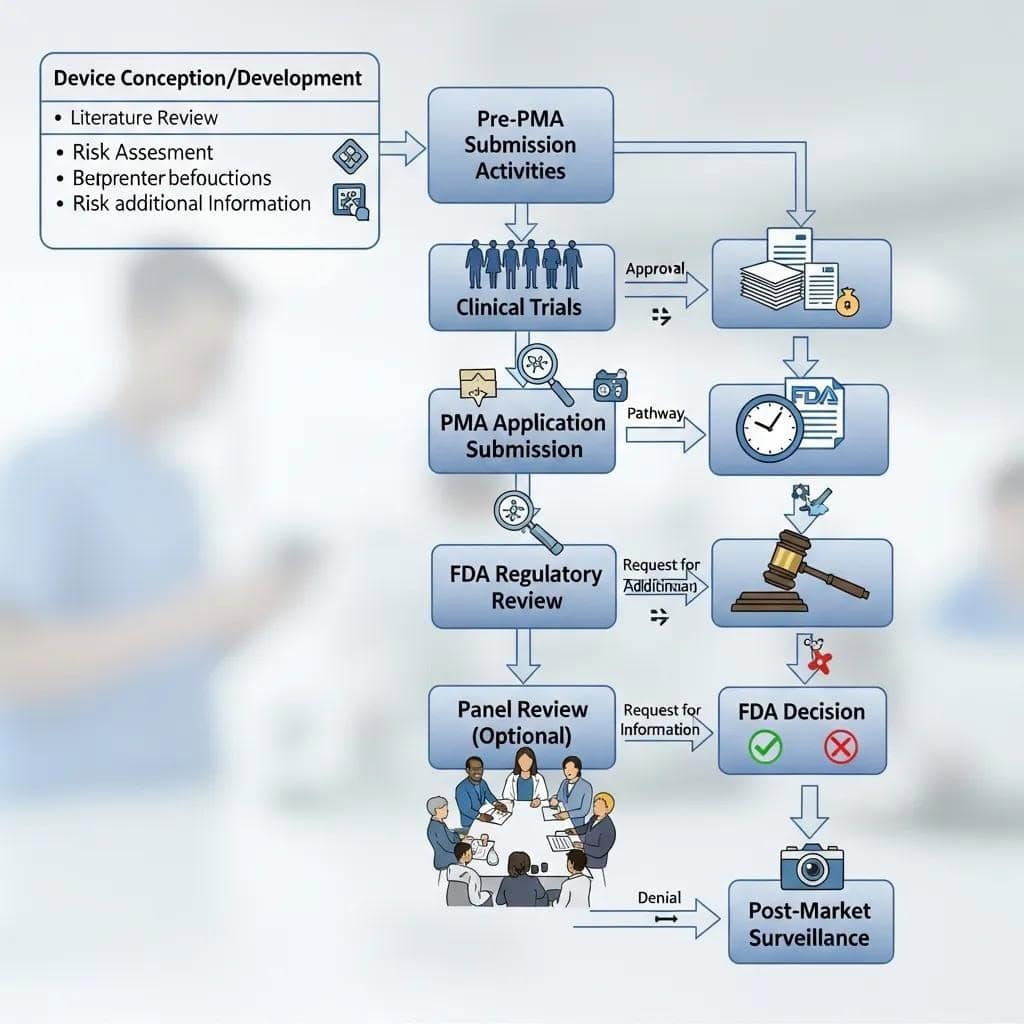
Clinical and Regulatory Requirements for PMA Approval
PMA applications require more than just a device. They must include IDE-supported pivotal trials with prospectively defined endpoints, robust statistical analysis plans, stringent safety monitoring, and often long-term patient follow-up. This is in addition to exhaustive documentation of manufacturing, sterilization, and device design.
The IDE submission itself secures permission to conduct investigational clinical testing. It must contain details on device manufacturing controls, investigator agreements, informed consent templates, and detailed monitoring plans. Pivotal trials, by definition, must be statistically powered to demonstrate the predefined primary endpoints with convincing efficacy.
CMC expectations are equally demanding. They require full descriptions of manufacturing processes, process validations, stability data, and rigorous supplier controls to ensure product reproducibility and safety. During the review, the FDA may convene an advisory committee for high-consequence devices.
Applicants should therefore be prepared to present integrated evidence and address complex questions regarding the device’s benefit-risk balance and long-term performance. Careful alignment between clinical endpoints, statistical plans, and manufacturing validation reduces the risk of PMA deficiencies. This integrated approach supports a streamlined review, balancing patient safety with timely market access for innovative, high-risk medical technologies.
CE Mark Medical Device Requirements Under EU MDR and IVDR
Obtaining a CE Marking under the European Union Medical Device Regulation (EU MDR) and In Vitro Diagnostic Regulation (IVDR) signifies conformity with European regulatory requirements. It obliges medical device manufacturers to produce comprehensive technical documentation, demonstrate compliance with general safety and performance requirements (GSPR), and, for many devices, engage a notified body for conformity assessment.
The MDR/IVDR frameworks significantly broadened the scope of regulation, tightened expectations for clinical evidence, and increased post-market surveillance obligations compared to the previous Directives. This makes robust clinical evaluation and proactive post-market clinical follow-up (PMCF) absolutely essential.
Classification rules under MDR/IVDR also differ from FDA classifications. Many devices have been reclassified into higher risk categories, directly impacting whether a notified body is required and which specific conformity assessment route applies. The following table summarizes key MDR/IVDR responsibilities and the typical evidence needed to support conformity assessments, assisting teams in planning their documentation and audits.
| MDR/IVDR Requirement | Who is Responsible / Typical Evidence | Purpose |
| Clinical Evaluation | Manufacturer / Clinical Evaluation Report (CER) | Demonstrate clinical safety and performance |
| Technical Documentation | Manufacturer / Technical File | Show conformity with General Safety & Performance Requirements |
| Notified Body Assessment | Notified Body / Audit Reports | Independent conformity assessment for higher-risk devices |
| Post-Market Surveillance (PMS) | Manufacturer / PMS Plan & Reports (including PMCF activities) | Ongoing safety monitoring and PMCF activities |
How the CE Marking Process Ensures Compliance with EU Medical Device Regulation
The CE Marking process in the EU follows a precise sequence. First, manufacturers must determine their device’s classification under the MDR/IVDR. Next, prepare comprehensive technical documentation and conduct a thorough clinical evaluation. Then, select the appropriate conformity assessment route. If required, undergo Notified Body audits and assessment before issuing a Declaration of Conformity and affixing the CE Mark.
The technical documentation must encompass a detailed device description, comprehensive risk management files, robust clinical evidence (CER), and clear post-market surveillance plans. Post-market clinical follow-up (PMCF) is frequently mandated, particularly for higher-risk devices. Timelines for CE certification are often affected by Notified Body capacity and the completeness of the technical file.
Common bottlenecks include insufficient clinical evidence, incomplete risk management documentation, or inadequate supplier oversight. A robust conformity plan typically involves early Notified Body engagement, clear clinical data strategies, and proactive PMS systems to meet MDR/IVDR expectations, thereby avoiding delays in CE certification. Preparing documentation that anticipates Notified Body questions significantly reduces iteration cycles, fostering efficient conformity assessment under the more stringent MDR/IVDR framework.
The Critical Role of Notified Bodies and Clinical Evaluation in CE Mark Approval
Notified Bodies are independent third-party organizations designated by EU member states to assess the conformity of higher-risk medical devices before they can be CE Marked. They conduct rigorous audits of quality management systems and meticulously review technical documentation, including Clinical Evaluation Reports (CERs). Their ultimate role is to issue certificates, which then permit the manufacturer to apply the CE Marking.
Selecting a Notified Body requires careful consideration of their scope, expertise with similar device types, and current audit timelines. Manufacturers should prepare for in-depth audits that scrutinize their QMS, supplier controls, and clinical evidence package. Clinical Evaluation Reports are particularly crucial.
They must systematically collect, appraise, and synthesize clinical data—drawing from published literature, clinical investigations, and post-market data—to demonstrate conformity with the GSPR. It is imperative that the evidence hierarchy is clearly presented, and any clinical data gaps are explicitly justified.
Practical preparation for Notified Body assessments includes meticulously organizing the technical file, demonstrating complete traceability from device requirements to verification activities, and maintaining robust, surveillance-ready PMS documentation. Thorough CERs and well-prepared Notified Body audits ultimately shorten certification timelines, minimize requests for additional information, and streamline the overall conformity assessment process.
ISO 13485 Certification: The Foundation of Medical Device Quality
ISO 13485 establishes the specific quality management system (QMS) requirements for the medical device industry. This international standard serves as a critical support mechanism for regulatory compliance, aligning processes for design control, supplier management, production, and post-market activities.
Achieving certification demonstrates a manufacturer’s commitment to consistent product quality. It provides a structured framework that maps effectively to both the FDA’s Quality System Regulation (21 CFR Part 820) and CE Mark expectations, rendering regulatory submissions and inspections far more predictable.
Key benefits of ISO 13485 certification include enhanced process control, clear traceability from requirements through verification and validation, strengthened supplier oversight, and formalized mechanisms for CAPA and risk management.
Collectively, these elements reduce nonconformances, improve overall product safety and efficacy, and support global market access. Implementing ISO 13485 early in product development embeds quality directly into all product lifecycle activities. This proactive approach often pre-empts many regulatory review issues by ensuring that necessary documentation and controls are robustly in place.

How ISO 13485 Supports Regulatory Compliance and Market Access
ISO 13485 provides standardized processes—including document control, design validation, supplier qualification, CAPA, and corrective action procedures. These directly align with the regulatory compliance expectations of both the FDA and EU authorities, making subsequent audits and submissions considerably more straightforward.
For example, a robust document control system ensures that test protocols, validation reports, and labeling are version-controlled, easily accessible, and readily auditable. Similarly, well-defined supplier management processes reduce variability in components and services, directly supporting the CMC requirements critical for PMA or 510(k) submissions.
Furthermore, a certified QMS formalizes post-market surveillance and feedback loops. This vital data directly informs product improvements and regulatory reporting, aligning seamlessly with the PMS and vigilance obligations mandated under MDR/IVDR legislation. Organizations that deeply integrate ISO 13485 into their product development lifecycle tend to encounter fewer review comments.
They can consistently demonstrate sustained regulatory compliance to both regulators and commercial partners, ultimately accelerating global market access. This operational alignment reduces friction during inspections and conformity assessments by establishing auditable, repeatable processes that regulators recognize as tangible evidence of reliable manufacturing and design practices.
Steps to Achieve and Maintain ISO 13485 Certification
Achieving ISO 13485 certification typically follows a structured roadmap. First, conduct a thorough gap analysis against the ISO requirements. Next, meticulously document all procedures and policies. Implement these processes across the organization, covering design control, supplier management, and CAPA. Conduct internal audits and management reviews to identify and address any nonconformities. Finally, undergo a certification audit by a recognized registrar.
While timelines vary, smaller organizations might anticipate 6–12 months for initial implementation and corrective activities before a successful certification audit. Maintaining certification requires continuous effort: periodic surveillance audits, ongoing internal audits, and meticulous documentation of continuous improvements. Integrating risk management and PMS into routine QMS activities can significantly reduce rework.
It supports long-term regulatory readiness. Practical tips include prioritizing document control, establishing a clear CAPA workflow, and ensuring all staff are thoroughly trained on their QMS responsibilities. This comprehensive, sustained approach creates lasting regulatory compliance. Following this stepwise plan ensures robust certification readiness and positions medical device manufacturers to confidently support regulatory submissions and strategic market expansion with a stable, high-quality platform.