Transform your innovative medical device concept into market reality with the right strategic partner
First, Let Us Explain How We Are Different So You Can Understand the Overall Strategy
Unlike traditional firms that treat medical device development like assembling farm equipment, we bring the precision of aerospace engineering to life-critical healthcare solutions. While others boast about “decades of experience” churning out commodity devices, our team combines extensive corporate leadership from industry giants with cutting-edge regulatory expertise and forward-thinking innovation strategies.
I bring over 15 years of engineering excellence across industry titans including Olympus, Terumo BCT, and Bolder Surgical, where he led breakthrough product launches from concept through FDA approval. My expertise spans everything from electrosurgical devices to novel respiratory systems, with a proven track record of transforming complex technical challenges into commercially successful medical devices.
Kelsey stands as one of the most accomplished regulatory strategists in the medical device industry, with an extraordinary portfolio spanning Medtronic’s global respiratory and monitoring divisions, leading IVD programs at Invitae Corporation, and directing regulatory affairs at Delfi Diagnostics. Her resume reads like a masterclass in regulatory excellence: over 12 FDA submissions in 12 months, successful PMA modules and De Novo pathways, and leadership of international regulatory teams across Boulder and Jerusalem. While others claim regulatory experience, Kelsey has actually architected the regulatory strategies that brought life-saving devices to market across orthopedics, patient monitoring, ventilators, and cutting-edge diagnostic platforms.
We don’t just follow established patterns—we anticipate regulatory evolution, leverage emerging technologies, and design with tomorrow’s standards in mind. While our competitors are still catching up to current regulations, we’re already designing for what’s coming next.

What Are the 5 Phases of Medical Device Development?
Understanding the medical device development process is crucial for making informed decisions about your project timeline, budget, and partner selection. Here’s how the industry’s most successful devices come to life:
Phase 1: Medical Device Concept Development
In Phase 1, we begin with a concept or identified clinical need. Our team of medical device designers and engineers conducts comprehensive market research to understand requirements and establish design inputs. We brainstorm multiple medical device concepts, then systematically narrow them down through feasibility analysis and stakeholder feedback.
Once the concept is developed, we provide medical device design services focused on proof-of-concept validation. Results are tested and validated through rapid prototyping technologies that quickly put prototype devices into end users’ hands for critical feedback on feasibility and usability. We iterate working models and support market research and focus groups as needed to ensure market-product fit.
Key deliverables: Market analysis, concept selection, initial prototypes, user feedback integration, feasibility assessment
Phase 2: Development & Design Controls
During Phase 2, we develop a comprehensive project plan with formal design controls compliant with FDA 21 CFR Part 820 and ISO 13485. We refine the conceptual medical device design through additional iterations and establish detailed engineering specifications and tolerances. Our engineers initiate hazard analysis using ISO 14971 risk management principles and begin development of robust test methods.
By the end of Phase 2, the design form has been selected, critical performance requirements tested, and design freeze achieved. This phase establishes the foundation for all subsequent verification and validation activities.
Key deliverables: Design controls documentation, engineering specifications, risk analysis, test method development, design freeze
Medical Device Development
- Market research & analysis
- Concept brainstorming
- Rapid prototyping
- User feedback integration
- Feasibility assessment
- Design control implementation
- Engineering specifications
- Risk analysis (ISO 14971)
- Test method development
- Design freeze
- Design verification testing
- Design validation studies
- Biocompatibility testing
- Clinical evaluations
- Packaging validation
- Process validation
- Supplier qualification
- Equipment qualification
- Shelf life studies
- Manufacturing documentation
- Regulatory approvals
- Production launch
- Staff training
- Inventory management
- Post-market surveillance
Phase 3: Verification & Validation
In Phase 3, we finalize comprehensive test methods for the medical device design. Design verification testing ensures performance meets all design input objectives and regulatory requirements. Design validation testing is performed under defined operating conditions—either actual or simulated use—on initial production units or equivalents.
We complete product packaging design and conduct biocompatibility testing in accordance with ISO 10993 standards. This phase also includes usability engineering studies and clinical evaluations as required by the device’s risk classification and intended use.
Key deliverables: Verification protocols and reports, validation testing, biocompatibility studies, packaging validation, clinical data collection
Phase 4: Manufacturing Scale-Up & Transfer
Our scale-up process includes comprehensive process validation, process capability studies, and sterility validation when applicable. Testing is performed on products built under full manufacturing-level documentation to ensure consistency and quality at production volumes.
Suppliers are qualified through rigorous evaluation processes, and production equipment is installed and qualified. Shelf life requirements are established and validated before Phase 4 completion. Final documentation is established, including labeling, instructions for use, and manufacturing procedures.
Key deliverables: Process validation, supplier qualification, equipment qualification, shelf life studies, manufacturing documentation
Phase 5: Full-Scale Manufacturing & Launch
During Phase 5, we secure all necessary regulatory approvals and implement comprehensive staff training for full production. Our deep understanding of compliance processes, including regulatory and design validation requirements, enables seamless design transfer and timely market launch.
We build initial inventory to support product launch and establish responsive supply chain management. Our Sales & Operations Planning services ensure efficient responses to demand forecasts and market fluctuations.
Key deliverables: Regulatory approvals, production launch, inventory management, post-market surveillance systems

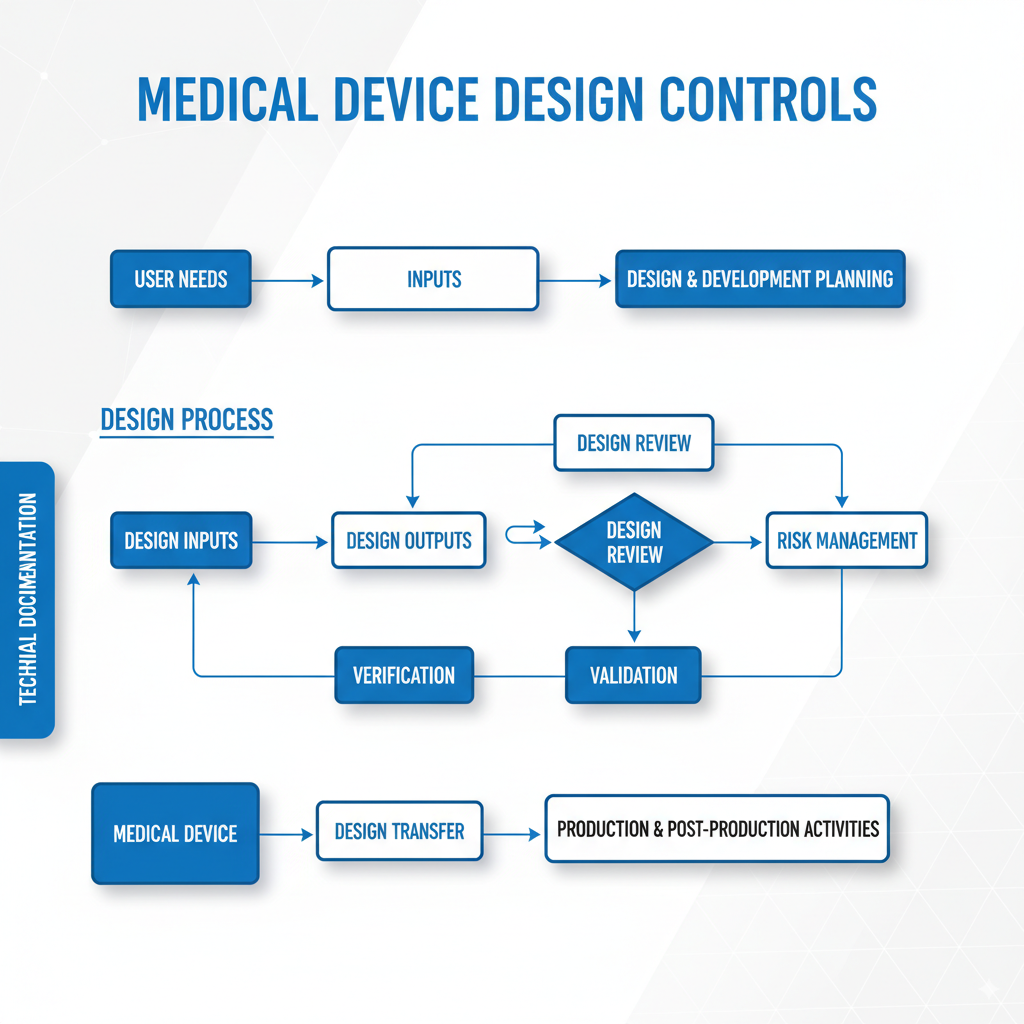
What Is the Design Process of a Medical Device?
The medical device design process follows a systematic approach governed by FDA design controls and international standards. This structured methodology ensures safety, efficacy, and regulatory compliance throughout development.
Design Inputs: Foundation of Success
Design inputs represent the essential requirements that define what the device must accomplish. These include user requirements, safety standards, regulatory constraints, performance specifications, and environmental conditions. Proper design inputs serve as the foundation for all subsequent design and development activities.
Critical design input categories:
- Performance requirements: What the device must achieve
- Safety requirements: Risk mitigation and user protection
- Regulatory requirements: FDA, EU MDR, and international standards
- User requirements: Clinical workflow integration and usability
- Environmental requirements: Operating conditions and durability
Design Outputs: Translating Requirements into Reality
Design outputs are the tangible results of the design process—specifications, drawings, risk analyses, protocols, and the device itself. Each design output must be traceable to specific design inputs and verified through testing.
Design Reviews: Ensuring Excellence Through Iteration
Design reviews provide systematic evaluation and refinement cycles throughout development. Cross-functional teams assess design progress against requirements, identify potential issues, and ensure design optimization before moving to the next phase.
Design Verification: Building It Right
Design verification confirms that the device meets specified design inputs through objective testing. This includes performance testing, safety evaluations, and regulatory compliance verification.
Design Validation: Building the Right Device
Design validation ensures the device meets user needs and intended uses under real-world conditions. This often involves clinical studies, usability testing, and end-user evaluations.
Design Controls: Comprehensive Documentation and Traceability
Design controls provide the framework for managing the entire design process. This includes maintaining comprehensive documentation, change control procedures, and traceability matrices linking inputs to outputs to verification activities.
Understanding Medical Device Classifications: Why It Matters for Your Bottom Line
Medical device classification directly impacts development timeline, cost, and regulatory requirements. Understanding these classifications early in your project enables accurate planning and budget allocation.
Class I vs. Class II vs. Class III: Timeline and Cost Implications
Class I Devices: The Express Lane
- Timeline: 6 months to 2 years
- Development cost: Under $1 million
- Examples: Surgical masks, tongue depressors, bandages
- Regulatory path: Simple registration and listing
- Key advantage: Fastest time to market
Class II Devices: The Sweet Spot
- Timeline: 1-3 years
- Development cost: $2-5 million
- Examples: Catheters, ventilators, patient monitors
- Regulatory path: 510(k) clearance demonstrating substantial equivalence
- Key advantage: Balanced risk-reward profile
Class III Devices: The Marathon
- Timeline: 3-7 years
- Development cost: $10-94 million
- Examples: Pacemakers, defibrillators, artificial hearts
- Regulatory path: Premarket Approval (PMA) with extensive clinical data
- Key advantage: Highest reimbursement and market protection
[IMAGE PLACEHOLDER: Medical device classification pyramid – Leonardo prompt: “Pyramid diagram showing medical device classifications, Class I at top, Class II middle, Class III at bottom, medical device icons for each level, professional blue gradient, clean business infographic style”]
Regulatory Pathway Selection Strategy
Choosing the optimal regulatory pathway requires deep understanding of predicate devices, regulatory precedents, and emerging FDA guidance. The wrong pathway selection can add years to development timelines and millions to costs.
510(k) Pathway Optimization:
- Predicate device analysis and selection
- Substantial equivalence demonstration
- Testing requirement minimization
- FDA communication strategy
De Novo Pathway Advantages:
- Novel device classification
- Lower risk category assignment
- Market exclusivity potential
- Regulatory precedent establishment
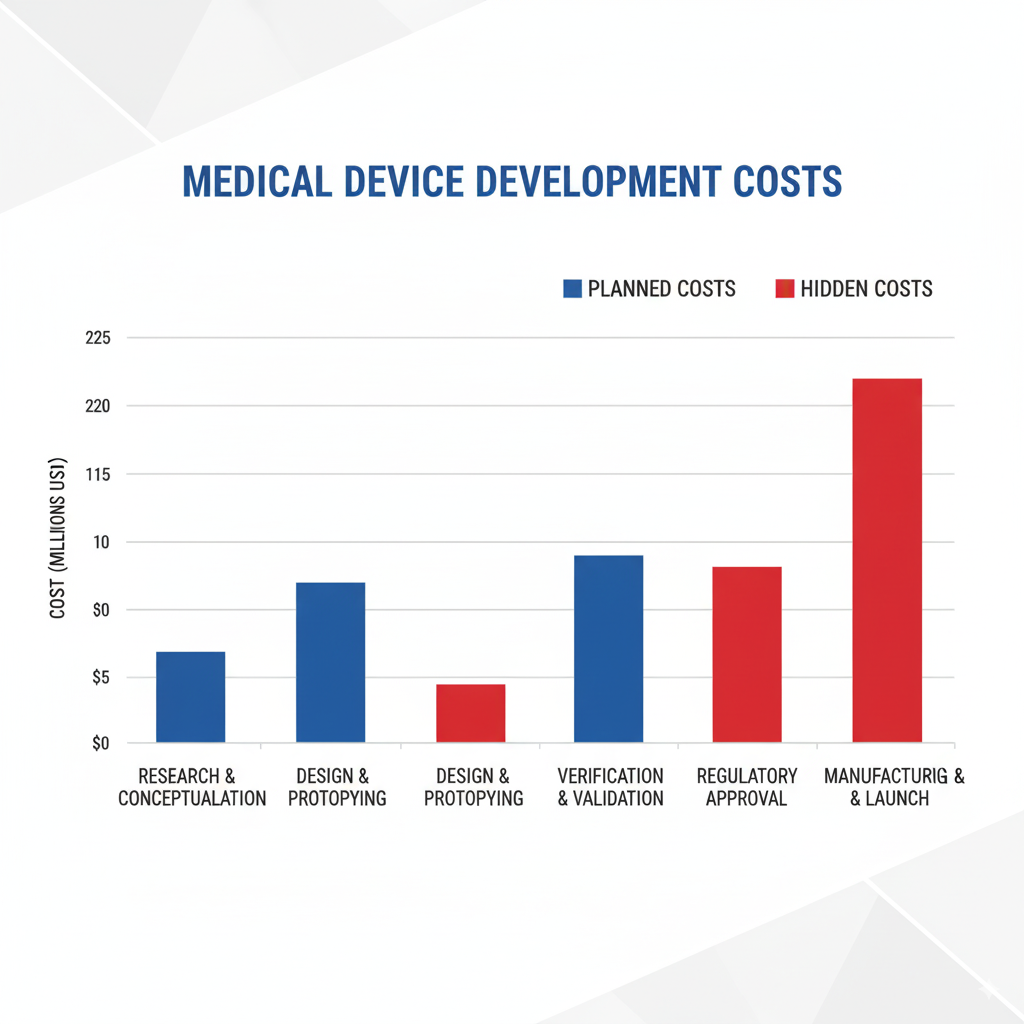
The Hidden Costs: Why 75% of Medical Device Startups Fail
Over 75% of new startups face challenges that can derail their projects, primarily due to inadequate planning and understanding of the complex medical device development landscape.
Common Pitfalls That Destroy Budgets
Documentation Gaps: The Silent Project Killer
Inadequate design controls documentation leads to regulatory delays, failed inspections, and expensive rework. Companies often underestimate the documentation requirements, resulting in scrambled efforts to compile missing records during FDA reviews.
Risk Management Failures: Costly Redesign Cycles
Poor upfront risk assessment using ISO 14971 principles leads to late-stage design changes when hazards are discovered during verification testing. These redesign cycles can double development timelines and budgets.
Design Control Deficiencies: Validation Nightmares
Insufficient design controls create traceability gaps between user requirements and final design outputs. This results in expensive validation studies that fail to demonstrate device safety and efficacy.
Regulatory Pathway Mistakes: The Million-Dollar Wrong Turn
Selecting the wrong regulatory pathway or inadequate FDA communication strategy can result in “Refuse to Accept” notices, requiring complete submission rework and adding 6-12 months to timelines.
Smart Investment Strategies for Device Development
Phase-Gate Funding Approach
Structure investment to align with development milestones, reducing risk exposure while maintaining development momentum. This approach enables course correction before major financial commitments.
Regulatory Consulting Integration
Early regulatory strategy development prevents costly late-stage surprises and optimizes submission timing. Invest in regulatory expertise upfront rather than scrambling during submission phases.
Quality System Early Implementation
Implementing ISO 13485 quality management systems early in development creates efficient documentation processes and prevents expensive retroactive compliance efforts.
Design Controls: Your Safety Net Against Catastrophic Failures
Design controls serve as the backbone of successful medical device development, providing structure, traceability, and regulatory compliance throughout the entire product lifecycle.
FDA 21 CFR Part 820 vs. ISO 13485: Which Path Is Right for You?
FDA 21 CFR Part 820: The U.S. Standard
- Prescriptive requirements with specific procedural mandates
- Focused on device safety and effectiveness
- Detailed labeling and packaging requirements
- Strong emphasis on change control and document management
ISO 13485: The Global Framework
- Risk-based approach with flexibility in implementation
- International harmonization enabling global market access
- Process-focused rather than prescriptive
- Easier integration with other management systems
Strategic Consideration: Many successful companies implement both standards simultaneously, enabling global market access while ensuring U.S. regulatory compliance.
Building a Bulletproof Design History File (DHF)
The Design History File serves as the complete record of design and development activities. A well-constructed DHF enables efficient regulatory submissions and protects against compliance issues.
Essential DHF Components:
- Design planning and procedures
- Design inputs and requirements
- Design outputs and specifications
- Design review records and decisions
- Verification and validation reports
- Risk management documentation
- Change control records
DHF Best Practices:
- Establish systematic filing procedures from project initiation
- Implement version control and document management systems
- Ensure traceability between all design elements
- Regular internal audits to identify gaps
- Cross-reference regulatory submission requirements
Manufacturing Readiness: From Prototype to Production Scale
Successful transition from development to manufacturing requires careful planning, process validation, and quality system implementation.
Design for Manufacturability (DFM): Avoiding the $2M Mistake
Design for Manufacturability principles integrated early in development prevent expensive manufacturing issues and enable cost-effective production scaling.
Critical DFM Considerations:
- Material selection: Optimize for both performance and manufacturability
- Process compatibility: Ensure design works with intended manufacturing methods
- Tolerance optimization: Balance performance requirements with manufacturing capabilities
- Assembly simplification: Minimize complexity and potential error sources
- Quality control integration: Design in inspection and testing capabilities
The $2M Mistake: Companies that ignore DFM principles often face expensive tooling changes, process modifications, and facility upgrades during scale-up. These costs, combined with launch delays, frequently exceed $2 million for complex devices.
Quality Management Systems That Actually Work
Effective quality management systems provide the foundation for consistent manufacturing and regulatory compliance.
ISO 13485 Implementation Strategies:
- Process mapping: Document all key processes and interactions
- Risk-based approach: Focus resources on highest-risk activities
- Continuous improvement: Establish metrics and improvement processes
- Supplier management: Integrate suppliers into quality system
- Employee training: Ensure competency and understanding throughout organization
Risk Management (ISO 14971) Integration:
- Hazard identification: Systematic evaluation of potential risks
- Risk estimation: Quantitative assessment of probability and severity
- Risk control: Implementation of mitigation measures
- Residual risk evaluation: Assessment of remaining risks
- Post-market surveillance: Ongoing monitoring and risk reassessment
Choosing the Right Medical Device Development Partner: A Strategic Framework
Selecting the optimal development partner significantly impacts project success, timeline, and cost. Use this framework to evaluate potential partners and make informed decisions.
Red Flags: When “Experience” Actually Means “Outdated”
Warning Signs to Avoid:
- Generic experience claims: “Decades of experience” without specific device examples
- Limited regulatory knowledge: Inability to discuss current FDA guidance or MDR requirements
- Outdated technology: Reliance on legacy design tools and methodologies
- Poor documentation practices: Inability to demonstrate robust design controls
- Limited quality system maturity: Absence of ISO 13485 certification or equivalent
Positive Indicators:
- Specific device portfolios: Demonstrated success with similar device types and classifications
- Current regulatory expertise: Active participation in FDA meetings and industry working groups
- Modern capabilities: Investment in current design tools, testing equipment, and facilities
- Quality system maturity: Established ISO 13485 systems with successful audit history
- Continuous learning: Evidence of ongoing training and capability development
Due Diligence Questions Every Executive Should Ask
Technical Capabilities Assessment:
- “Show us three devices similar to ours that you’ve successfully brought to market.”
- “What’s your approach to design controls and how do you ensure FDA compliance?”
- “How do you handle risk management throughout the development process?”
- “What testing capabilities do you have in-house vs. outsourced?”
- “How do you approach design for manufacturability?”
Regulatory Expertise Evaluation:
- “What’s your experience with our device’s specific regulatory pathway?”
- “How do you stay current with changing FDA guidance and regulations?”
- “Can you provide examples of successful regulatory submissions for similar devices?”
- “What’s your relationship with FDA and how do you manage agency communications?”
- “How do you handle international regulatory requirements?”
Project Management and Communication:
- “What project management methodologies do you use and why?”
- “How do you handle scope changes and timeline adjustments?”
- “What’s your communication cadence and reporting structure?”
- “How do you manage intellectual property and confidentiality?”
- “What happens if key team members leave during our project?”

Timeline Expectations: Realistic Planning for Device Development
Understanding realistic timelines enables accurate project planning and stakeholder management throughout the development process.
Factors That Accelerate or Derail Your Project
Timeline Accelerators:
- Clear requirements definition: Well-defined user needs and specifications
- Appropriate regulatory pathway: Optimal route selection based on device characteristics
- Experienced team: Proven track record with similar devices and regulations
- Robust project management: Structured approach with clear milestones and accountability
- Early regulatory engagement: Proactive FDA communication and guidance seeking
Common Derailment Factors:
- Scope creep: Uncontrolled feature additions and requirement changes
- Regulatory surprises: Unexpected pathway complications or additional requirements
- Design control gaps: Inadequate documentation requiring extensive rework
- Testing failures: Inadequate initial design leading to multiple test iterations
- Supplier issues: Unqualified suppliers or component availability problems
Managing Stakeholder Expectations Throughout Development
Executive Communication Strategy:
- Monthly executive summaries: High-level progress and risk updates
- Quarterly business reviews: Detailed progress assessment and planning
- Milestone celebrations: Recognition of key achievements and team contributions
- Risk communication: Transparent discussion of challenges and mitigation strategies
Investor Relations Management:
- Phase-gate reporting: Progress updates aligned with funding milestones
- Regulatory milestone tracking: Clear communication of submission timing and results
- Market preparation updates: Commercial readiness and launch planning progress
- Competitive landscape monitoring: Market changes and strategic implications
Why Forward-Thinking Companies Choose Specialized Expertise Over Generic Experience
The medical device industry demands specialized knowledge, current regulatory understanding, and proven execution capabilities. Generic consulting firms simply cannot match the depth and breadth of expertise required for successful medical device development.
The Specialization Advantage:
- Deep regulatory knowledge: Current understanding of FDA requirements and emerging guidance
- Industry relationships: Established connections with key suppliers, testing labs, and regulatory consultants
- Proven methodologies: Refined processes optimized for medical device development
- Risk mitigation: Experience-based understanding of common pitfalls and prevention strategies
- Efficiency optimization: Streamlined approaches that reduce timeline and cost
Beyond Experience: The Innovation Factor
True differentiation comes not just from experience, but from the ability to anticipate and adapt to evolving industry requirements. Our team combines extensive corporate experience with entrepreneurial agility, enabling innovative solutions that address tomorrow’s challenges today.
Ready to transform your innovative medical device concept into market reality? Partner with experts who understand both today’s requirements and tomorrow’s opportunities.
Transform your innovative medical device concept into market reality with partners who understand both today’s requirements and tomorrow’s opportunities. Contact us to discuss how our unique combination of corporate expertise and regulatory mastery can accelerate your path to market.
About the Authors:
Eric Klumper brings over 15 years of medical device engineering expertise across industry leaders including Olympus, Terumo BCT, and Bolder Surgical. His proven track record includes successful product launches from concept through FDA approval across diverse medical device categories.
Kelsey Lee stands as one of the industry’s most accomplished regulatory strategists, with an extraordinary portfolio spanning Medtronic’s global operations, IVD program leadership at Invitae Corporation, and regulatory affairs direction at cutting-edge diagnostic companies. Her expertise encompasses over 12 FDA submissions in 12 months and successful navigation of complex regulatory pathways.
Ready to Get Started?
Transform your medical device vision into reality with proven expertise and forward-thinking innovation. Contact our team to discuss your project and discover how our unique approach can accelerate your path to market success.
[Contact Us Today] [Download Our Capabilities Overview] [Schedule a Consultation]